
CLN - Feature

Much to the dismay of many laboratorians nationwide, the Food and Drug Administration (FDA) on May 6 issued its final rule on laboratory developed tests (LDTs). The rule stakes claim to regulation of laboratory testing services as in in vitro diagnostics (IVD) devices defined by the Federal Food, Drug, and Cosmetic Act. Although the agency will not immediately require FDA clearance or approval of all LDTs, and defined several carveouts for “enforcement discretion,” even tests that fall into these special categories will require new documentation, compliance systems, and reporting to the FDA that laboratories have never done.
The FDA spelled out a four-year plan to implement the changes. However, it is likely that some of the provisions may wind up altered because of at least one lawsuit challenging the FDA, the presidential election in November, or actions from Congress, experts say.
“Clinical laboratories currently have no infrastructure to do what the FDA wants us to do, and 4 years is too aggressive a timetable,” said Dennis Dietzen, PhD, DABCC, FADLM, professor of pathology and immunology at Washington University School of Medicine and medical director of laboratory services at St. Louis Children’s Hospital. Even when using so-called “grandfathered” tests, labs still have to comply with quality documentation records and registration and reporting of errors, he said, and some patients, particularly newborns whom he designs tests for, could face reduced access to testing or face delays: “I think we’re all hopeful that this is going to get changed, delayed, or annulled in some way.”
The FDA had considered an LDT to be an in IVD intended for clinical use and designed, manufactured, and used within a single laboratory certified under CLIA to perform high-complexity testing, said Elizabeth Hillebrenner, associate director for scientific and regulatory programs at FDA’s Center for Devices and Radiological Health, during a May 14 FDA webinar to explain the rule.
For years, FDA maintained enforcement discretion, she said, as LDTs were mostly performed in small volumes by laboratories serving their local communities, employed manual techniques, and were intended for use in diagnosing rare diseases. Today, labs offer IVDs as LDTs more often in high volume for large, diverse populations; increasingly rely on high-tech or complex instrumentation and software; and are used in laboratories outside a patient’s healthcare setting, she said. Additionally, they are more commonly performed by independent labs, using instruments or other components not legally marketed for clinical use, and then marketed nationwide.
“As a result of these evolutions in the testing landscape, FDA has long recognized the need for a change in the agency’s general enforcement discretion approach for LDTs,” Hillebrenner said. “We believe this rule will better protect the public health by helping to assure the safety and effectiveness of IVDs offered as LDTs while also accounting for other public health considerations, such as patient access and reliance.”
The rule, published May 6 and officially taking effect July 5, requires laboratories to comply with a litany of requirements over a four-year “phase out” period, explained Jonathan Genzen, MD, PhD, chief medical officer and senior director of government affairs at ARUP Laboratories, in a May 23 webinar hosted by ARUP (See chart).
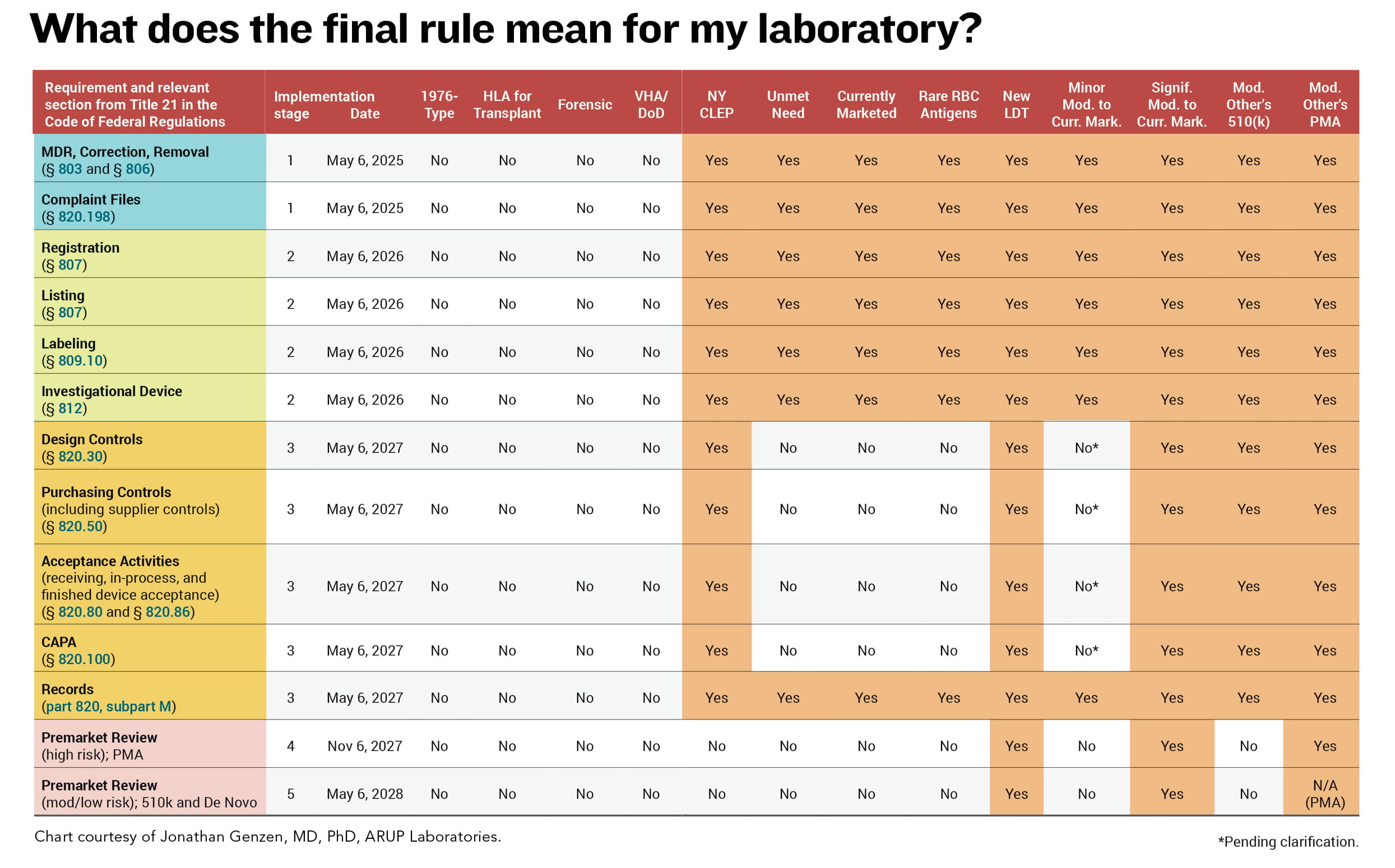
In one year — by May 6, 2025 — laboratories need to comply with stage 1 requirements, including medical device reporting, requirements related to device correction and removal from the market, and maintaining files of complaints about assays. By May 6, 2026, laboratories need to comply with stage 2 requirements for LDTs including registration and listing, labeling, and investigational use.
“Labeling is something that I would urge laboratories to start reading about early on to make sure that they can comply within that two-year timeframe,” Genzen said. “There’s a lot of important information in those documents and labs would need to compile that information according to FDA requirements. And if your LDTs are being used in investigational studies by other individuals, there may be additional requirements that the laboratory must now follow, described in the Code of Federal Regulations.”
Labeling also includes information on unique device identifiers (UDIs) — requirements specifically related to labeling that help laboratorians trace back each individual component or reagent, he added. “This is going to be very challenging for laboratories that are not physically manufacturing kits but assembling these items as test systems. That’s why I recommend people start studying labeling early on.”
By May 6, 2027, laboratories need to comply with stage 3 quality system requirements other than complaint files. “This is also a big deal,” Genzen said, and another area that labs should look into early “because there are significant requirements related to how you do your design work related to LDTs and how you document information that is probably different than the way that you are documenting information currently.”
By November 6, 2027 (stage 4) and May 6, 2028 (stage 5), laboratories need to submit certain assays for premarket review. Stage 4 relates to high-risk IVDs requiring premarket authorizations, and stage 5 relates to moderate- and low-risk IVDs that would require the 510K or de novo review or clearance.
There are several areas in which the FDA said it intends to continue enforcement discretion, Genzen and Hillebrenner said: “1976-type” LDTs, or manual tests that involve no automated preparation or interpretation, such as immunohistochemistry tests or tests that use staining antibodies and general purpose reagents for cytology, hematology, and bacterial infections; human leukocyte antigen tests specifically for transplant; forensic tests intended solely for law enforcement/forensic purposes; and LDTs developed and performed within Veterans Administration hospitals and the Department of Defense.
Exclusions also generally will apply to the following: LDTs approved by the New York State Department of Health’s Clinical Laboratory Evaluation Program; LDTs marketed prior to May 6 that either have not been modified or modified in certain limited ways; nonmolecular antisera LDTs for rare red blood cell antigens; and LDTs developed and performed by a laboratory integrated within a healthcare system for an unmet need of patients receiving care within the same healthcare facility. This last exception is an area hospital laboratories will carefully need to scrutinize, Genzen said.
For example, FDA does not consider “unmet need” to apply to patients being treated at an affiliated hospital with a different corporate ownership than the laboratory. The policy also is limited to LDTs ordered by a healthcare practitioner on the staff or who has credentials and privileges at a facility owned and operated by the same healthcare system employing the laboratory director and performing the LDT.
“They’ve created a very restrictive definition of unmet needs that actually is not going to be applicable in many settings,” he cautioned. As detailed as the rule is, there are still many unanswered questions, such as what to do about tests scheduled to be released before the stages set in, Genzen told CLN: “We will need much more specific information.”
ADLM has outspokenly opposed this rule since a draft was released in October 2023, advocating for the FDA to withdraw their proposed rule and work with the laboratory community, patients, and Congress to update CLIA standards, which since 1988 have been the mechanism for regulating LDTs.
“ADLM agrees that increases in the number and complexity of LDTs may necessitate a review of the regulations governing these critically important clinical testing services … [but] is concerned that extending FDA oversight to LDTs will duplicate the existing regulatory structure, diverting limited laboratory resources from the provision of care to new, unnecessary administrative requirements,” ADLM president Octavia Peck Palmer, PhD, FADLM, said in written testimony before the House Energy and Commerce Subcommittee on Health in March 2024.
“The FDA states that laboratories that develop LDTs are medical device manufacturers and must be subject to the same requirements,” Peck Palmer said. “The agency assumes that hospitals, small community testing facilities, and other providers can afford the technical and administrative staff necessary to perform the studies, file the submissions, provide supplemental information, and continue an ongoing dialogue with the FDA to gain agency clearance or approval of an LDT. We are concerned that the costs associated with this duplicative regulatory structure will be significant for many healthcare facilities, forcing them to discontinue or scale back these services.”
Other laboratorians have expressed alarm, too.
Dietzen said the unmet need clause may be helpful for laboratories like his that build tests when no FDA-approved alternative exists, Dietzen said, “But the way that we have to build these is going to be drastically different than the way we do it today under CLIA.” The FDA has thrown additional requirements on top of those from CLIA, he added, “and in some cases, they are likely to be duplicative or maybe even contrary to one another.”
“I’m concerned about the impact on diagnostic innovation going forward, that the … financial burden of introducing new tests will increase significantly,” Genzen said. The FDA acknowledged in their regulatory impact analysis and final rule preamble that this may disproportionately affect smaller labs, he said: “Those are really important settings.”
Many of the negative impacts will not appear until the second year or later after the rule’s release, when the requirements hit and labs will decide whether to discontinue or outsource some testing, he added. While some question the FDA’s right to oversee LDTs, the agency did cite legal cases in their claim for this authority, said James Boiani, JD, an attorney with Epstein Becker Green in Washington, D.C. However, this has not ever been decided by courts, he noted, “so it’s certainly open to challenge.”
The American Clinical Laboratory Association (ACLA) and its member company, HealthTrackRx, already took action, filing a lawsuit on May 29 in the U.S. District Court for the Eastern District of Texas against the FDA to challenge its authority. Named as codefendants are the Department of Health and Human Services, HHS Secretary Xavier Becerra, and FDA Commissioner Robert Califf. ACLA has worked collaboratively with Congress, FDA, and others on legislation to establish “an appropriate regulatory framework for diagnostics,” according to an ACLA news release announcing the suit. “Rather than continue that dialogue, FDA chose instead to act unilaterally and impose an ill-fitting device regime on laboratory testing services.”
“If it is not vacated, FDA’s unprecedented final rule will have devastating and far-reaching consequences not only for the nation’s clinical laboratories, but also for the nation’s entire healthcare system, including the millions of vulnerable patients who depend on the essential clinical testing services that laboratories provide,” the complaint states. It includes declarations from Genzen as well as executives from Labcorp, Quest Diagnostics, and Mayo Clinic Laboratories.
It’s likely that either additional laboratories will sue or Congress will somehow get involved, potentially as part of FDA’s periodic negotiations for funding for its medical device user fee program, Boiani said. “There are going to be changes and revisions and challenges, probably in the courts and also on Capitol Hill, as this works its way forward to implementation.”
There’s a very high chance that this is not going to play out exactly as written, said Bruce Quinn, MD, PhD, a diagnostic consultant with Bruce Quinn & Associates, for several reasons. One, there are limited staff at the FDA and in the industry trained on proper regulatory submissions, and gathering many times that number will take time. This or additional potential lawsuits could tie up progression of the rule for at least a year or two. And, with the November presidential election on the horizon, a potential change in administration could upend things as well.
Meanwhile, as a first step, start assessing what the impact of the rule will be on your laboratory, Boiani advised. See what the new regulations will require and try to map out how it’s going to impact your operations, so you have a sense of what sort of investments you may need to make if the rule goes forward, and whether it’s worth challenging, he said. Consider the costs of the quality system and adverse event reporting requirements, registration fees, and whether you need any additional licenses from your state, as well as any Sunshine Act reporting payments to physicians.
Genzen concurred: “I strongly recommend people use this time to increase their own understanding of quality system requirements, labeling requirements, and even investigational use requirements if they have clinical services that are using their tests for research or investigational purposes.”
Additionally, Boiani said, follow the guidances issued by the FDA and submit comments if you see a problem. The agency did adjust some pieces from the proposed to the final rule based on comments. “FDA has people with experience in the industry, but labs that are living this day to day are going to know the practical problems with what might be proposed,” he said.
Some laboratories will go to one of two extremes, Quinn said. They'll ignore the rule completely, “which might seem rational, because something will probably happen. The other extreme would be devoting half of your cash flow trying to hire people and meet these rules. I really don’t know what the answer is.”
Meanwhile, “All of us are going to have to sit tight and watch how this plays out,” Dietzen said. FDA intends to hold additional webinars on specific aspects of the final rule, targeted enforcement discretion policies, and other matters applicable to IVDs including LDTs, Hillebrenner said. ADLM launched a series of webinars on LDTs available on www.myadlm.org.
Karen Blum is a freelance medical/science writer in Owings Mills, Maryland. +Email: [email protected].