
Academy of Diagnostics & Laboratory Medicine - Guidance Document
Guidance Documents are brought to you by the

Yusheng Zhu
Department of Pathology and Laboratory Medicine,
Department of Pharmacology
Pennsylvania State University College of Medicine
Hershey, PA, USA
Sarah Feldman
Departments of Obstetrics, Gynecology, and Reproductive Biology
Brigham and Women’s Hospital, Harvard Medical School
Boston, MA, USA
Shuk On Annie Leung
Department of Obstetrics and Gynecology
McGill University Health Centre
McGill University
Montreal, QC, Canada
Michael H. Creer
Department of Pathology and Laboratory Medicine
Pennsylvania State University College of Medicine
Hershey, PA, USA
Joshua Warrick
Department of Pathology and Laboratory Medicine
Pennsylvania State University College of Medicine
Hershey, PA, USA
Nicole Williams
Department of Pathology and Laboratory Medicine
Pennsylvania State University College of Medicine
Hershey, PA, USA
Stephen Mastorides
Department of Pathology and Laboratory Medicine Service
James A. Haley Veterans’ Hospital
Tampa, FL, USA
Cervical cancer is a group of invasive epithelial neoplasms of the cervix, all of which have metastatic potential. These comprise 70% squamous cell carcinoma and 25% adenocarcinoma, with the remainder rare tumors, such as small cell carcinoma (1). The vast majority of cervical cancers are driven by infection with high-risk human papilloma virus (hrHPV), most notably HPV types 16 and 18, which are responsible for about 70% of cervical cancers (1). Human papilloma virus (HPV) is a double-stranded DNA virus with over 200 known genotypes. In addition to types 16 and 18, other clinically relevant high-risk types include 58, 33, 45, 31, 52, 35, 59, 39, 51, 56, 66, and 68 in order of worldwide frequency from high to low. Several biological steps must take place for infection with hrHPV to progress to cervical cancer (2). The earliest and most obvious is HPV acquisition, which is often spontaneously cleared (3). This can be seen histologically as the koilocytotic atypia characteristic of low-grade squamous intraepithelial lesions (4). If HPV infection persists, viral DNA integrates into the host genome, inducing expression of high levels of oncogenic viral proteins, such as E6- and E7-encoded oncoproteins, which facilitate degradation of the host tumor suppressor proteins p53 and RB1, respectively (3). These are seen histologically in either the extensive basaloid atypical characteristic of high grade squamous intraepithelial lesions or the atypical noninvasive glands of adenocarcinoma in situ (4). Over time, these cells acquire somatic driver mutations and invade. The most common somatic mutations involve members of the PI3K/AKT pathway, specifically activating mutations in PIK3CA and copy number losses or inactivating mutations of PTEN, seen in both squamous cell carcinoma and adenocarcinoma (5, 6). As the disease progresses, invasive cervical cancers are capable of local invasion as well as distant metastasis and patient mortality.
Histopathologically, the precursor lesions of squamous cell carcinoma of the cervix are termed cervical intraepithelial neoplasia (CIN), which divides cervical cancer precursors into 3 groups: CIN 1, 2, and 3, corresponding to mild dysplasia, moderate dysplasia, and severe dysplasia/carcinoma in situ, respectively. For exfoliative cytology specimens, cervical cancer precursors are classified as low-grade squamous intraepithelial lesion (LSIL) for lesions histopathologically classified as koilocytotic atypia and CIN 1 and high-grade squamous intraepithelial lesion (HSIL) for lesions called CIN 2 and CIN 3 in histopathology. For histopathological reporting, it has been suggested using LSIL (CIN1) and HSIL (CIN 2 and CIN 3) (4) and both terminology systems are currently in use. The 2019 American Society for Colposcopy and Cervical Pathology (ASCCP) guidelines recommend that the histopathology report should include CIN 2 or CIN 3 qualifiers, that is, HSIL (CIN 2) and HSIL (CIN 3) (7). Approaches for cervical cancer screening include primary cervical hrHPV testing, co-testing of hrHPV and cervical cytology, and cytology screening alone. These approaches have variable sensitivity and specificity, which will be detailed in the later sections (8).
In addition to screening, cervical cancer tests are used in surveillance as well as diagnosis of cervical cancer. It is important to distinguish between screening, surveillance, and diagnostic testing. Screening refers to testing for disease among individuals who are asymptomatic and have not been tested previously or have normal prior results (i.e., low risk). Surveillance is the interval testing among individuals who had a prior abnormal result, with or without treatment. Recent evidence indicates that an individual’s risk of developing cervical precancer or cancer can be estimated using current screening test results and previous screening test and biopsy results, while considering personal factors such as age and immunosuppression (7). These data form the basis of the 2019 ASCCP risk-based management consensus guidelines for abnormal cervical cancer screening tests and cancer precursors, which will be discussed in later sections (7). When an individual’s history is unknown, that individual’s risk falls somewhere in between screening and surveillance. It is important to note that an unknown history is itself a risk factor for development of cervical precancer and cancer (9). Finally, diagnosis refers to testing including colposcopy and biopsy when an individual presents with symptoms (e.g., bleeding, discharge, pain). In addition to biopsy for histologic diagnosis, note that cytology and/or HPV testing may also be used by clinician as part of a comprehensive work-up to guide management. The distinction between these three categories (screening, surveillance, and diagnosis) is important because, although similar tests might be utilized, the subsequent interpretation of risk to guide management is different.
This guidance document introduces currently available cervical cancer screening tests, testing strategies, and the most recently updated screening guidelines as well as risk-based management guidelines. In addition, we propose a report template for HPV and cervical cancer detection to facilitate interpretation of testing results and clinical decision-making.
Currently, cervical cancer screening tests include HPV testing and cervical cytology in clinical settings. Recently, it has been proposed that self-collected vaginal specimens are suitable for HPV testing, although the Food and Drug Administration (FDA) has not yet approved any self-collection methods.
HPV testing may be used alone for primary hrHPV screening or in conjunction with cervical cytology as part of a co-testing strategy, which will be discussed in detail in the “Screening Strategies” section. There are currently 5 FDA-approved HPV molecular assays (10–14).
The FDA approved the assay in 2011 for reflex HPV testing and co-testing with cytology. In 2014, it was approved for primary cervical cancer screening but only on Hologic ThinPrep specimens (see the following “Cervical Cytology Test” section). The DNA real-time qPCR-based assay targets the L1 gene of HPV. It covers 14 high-risk types (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, and 68) with genotyping of 16 and 18. The beta-globin gene serves as an internal control. The sensitivity for detecting CIN 2/3 ranges from 90.5% to 97% and the specificity ranges from 13% to 67.6% (15–17).
(b) The BD Onclarity™ HPV Assay (Becton, Dickinson and Company)
The FDA approved this assay in 2018 for reflex HPV testing and co-testing with cytology as well as primary cervical cancer screening but only on SurePath Specimens (see the following “Cervical Cytology Test” section). The DNA PCR-based assay targets E6/E7 genes. It covers 14 high-risk types including 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, and 68 with genotyping of 16, 18, and 45. The beta-globin gene serves as an internal control. The sensitivity for detecting CIN2/3 ranges from 94% to 98% and the specificity ranges from 17% to 31% (17–19).
The FDA approved this assay in 2001 for reflex HPV testing and co-testing with cytology. The DNA signal amplification (non-PCR) assay utilizes a full genome probe. It covers 13 high-risk types (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, and 68). There is no built-in internal control. The sensitivity for detecting CIN 2/3 ranges from 80.8% to 98% and the specificity ranges from 21% to 70.6% (10, 16–20).
(b) Cervista HPV HR Assay (Hologic Inc.)
The FDA approved this assay in 2009 for reflex HPV testing and co-testing with cytology. The DNA signal amplification (non-PCR) assay targets L1, E6, and E7 genes. Cervista HPV HR assay covers 14 high-risk types (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, and 68). The Cervista HPV 16/18 assay tests HPV 16 and 18 only. The HIST2H2BE gene serves as an internal control. The sensitivity for detecting CIN2/3 ranges from 77% to 92.8% and the specificity ranges from 44.2% to 72.7% (21–23).
(c) Aptima HPV Assay (Hologic Inc.)
The FDA approved the assay in 2011 for reflex HPV testing and co-testing with cytology. The mRNA transcription-mediated amplification assay targets E6/E7 genes. It covers 14 high-risk types (16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, and 68) with separate genotyping of 16, and 18/ 45 by the Aptima 16,18/45 genotype assay. The HPV16 E6/7 transcript serves as an internal control. The sensitivity for detecting CIN2/3 ranges from 87.5% to 98% and the specificity ranges from 30% to 78% (10, 15, 17, 19, 24).
Cervical cytology screening, also known as the Pap smear test, involves the direct sampling of the transformation zone between the ectocervix and endocervix. The traditional Pap test involves collecting cells from the vagina or cervix, smearing them onto a slide at the patient bedside, and evaluating the slide in the laboratory under a microscope. A significant advance in cervical cancer screening is the introduction of liquid-based cytology (LBC). Currently, LBC is utilized in over 90% of Pap tests in the United States and has higher sensitivity for high-grade lesions than conventional smears with a lower false negativity rate (25–28). LBC was first approved by the FDA in 1996 with the ThinPrep® Pap test (Hologic Inc.). The FDA approved a second test in 1999, the BD SurePath™ Pap test (BD Diagnostic).
The ThinPrep® Pap test sample is collected by a clinician with a plastic spatula and an endocervical brush or a Cervex-Brush Combi device (a broom-like device with an integrated endocervical sampler) and rinsed in a ThinPrep vial prefilled with a methanol-based fixative (PreservCyt). The vial is sent to the laboratory for processing on the ThinPrep Processor, an automated slide preparation unit that uses a liquid-based vacuum filtration method to disperse, filter, and transfer the specimen onto a slide using air pressure for adherence resulting in a uniform monolayer of cells. The residual specimen is available for other diagnostic tests, for example, HPV testing (26, 27).
The SurePath™ Pap test sample is collected by a clinician using a broom-like device with a detachable head. The sample is placed in a collection vial with an ethanol-based fixative (CytoRich) and sent to the laboratory for processing. The cells are centrifuged, suspended within a sucrose density gradient, and transferred to slide via gravity for adherence in a monolayer. The residual specimen is available for other diagnostic tests, for example, HPV testing (26, 27).
Both ThinPrep and SurePath Pap tests are approved for primary screening by automated imagers. The ThinPrep Imaging System (TIS, Hologic Corp.) is used with ThinPrep slides, and the FocalPoint GS Primary Screening System (Focal Point GS, BD Diagnostics) can be used with SurePath and conventional Pap tests. The automated imagers have slightly increased sensitivity over manual screening alone; however, there is a slight decrease in specificity (29–32).
Evaluation of slides by automated screening or manual screening by a cytotechnologist or cytopathologist is considered primary review. All abnormal cervical Pap smears must have a secondary review by a cytopathologist. The reporting of results follows the Bethesda System for Reporting Cervical Cytology (Table 1) (33).
The spectrum of lesions in the cervix caused by HPV ranges from premalignant dysplasia to invasive carcinoma. Low-grade dysplasia or LSIL in cytology may be indicative of HPV infection that can be transient with regression within 2 years (34). Cytomorphologic changes of LSIL in Pap test are similar to those identified as CIN 1 in cervical tissue biopsies. Changes of LSIL can range from viral cytopathic change (koilocytosis) to morphologic changes of low-grade dysplasia. A Pap test with atypical changes involving squamous cells that fall short of criteria for LSIL can be reported as atypical squamous cells of undetermined significance (ASC-US). The ASC-US/LSIL ratio is a laboratory quality indicator and can highlight ASC-US overuse.
|
Table 1. The Bethesda system for reporting cervical cytology diagnostic categories. |
||
|
Diagnostic category |
Comments |
|
|
Unsatisfactory |
Inadequate |
|
|
cellularity, |
||
|
Obscuring |
||
|
inflammation or |
||
|
blood |
||
|
Negative for intraepithelial |
Nonneoplastic |
|
|
lesion or malignancy (NILM) |
(tubal metaplasia, |
|
|
pregnancy |
||
|
changes, atrophy), |
||
|
Reactive changes, |
||
|
Organisms/viral |
||
|
cytopathic |
||
|
changes |
||
|
ASC-US |
||
|
ASC-H |
||
|
Squamous intraepithelial lesion |
HPV cytopathic changes |
|
|
-LSIL |
||
|
-HSIL |
||
|
Squamous cell carcinoma |
||
|
Glandular cells |
||
|
-Atypical |
||
|
||
|
||
|
||
|
-Atypical |
||
|
||
|
||
|
-Adenocarcinoma in situ |
||
|
-Adenocarcinoma |
||
|
Other malignancy |
Metastatic tumors, |
|
|
sarcoma, |
||
|
neuroendocrine |
||
|
tumors, etc. |
||
|
NOS, not otherwise specified. |
||
The cytomorphology of HSIL is similar to CIN 2 and CIN 3 in tissue biopsy. Squamous cells with high-grade dysplasia are smaller than those with low-grade dysplasia. They have high nuclear to cytoplasmic ratio and marked nuclear membrane irregularity and can have nuclear hyperchromasia. Atypical changes that fall short of criteria for HSIL can be reported as atypical squamous cells—cannot exclude high-grade squamous intraepithelial lesion (ASC-H).
Squamous cell carcinoma is the most common malignancy of the cervix. Tissue architecture is not present in a cytology sample; however, other malignant features are present. Tumor cells can have similar cytomorphology as those seen in HSIL; however, these cells also may have increased pleomorphism and dense eosinophilic cytoplasm in keratinizing squamous cell carcinoma. Additionally, an associated tumor diathesis comprised of necrotic debris and degenerated blood clings to cells in liquid-based cytology.
Cervical cytology is a screening test for squamous lesions; however, atypical glandular cells (AGC) and changes suggestive of glandular malignancies can also be identified. There is lower sensitivity for glandular lesion detection by cytology due to several issues, including cellular degeneration, interpretation, and sampling. AGC can be endocervical or endometrial; however, it may not be possible to identify the origin based on cytology alone. AGC in Pap test samples may correlate to reactive inflammatory lesions, extension of squamous dysplasia into endocervical glands, in situ, or invasive adenocarcinoma in tissue biopsy specimens. Cytomorphologic changes of atypia include nuclear enlargement with overlapping, increased nuclear to cytoplasmic ratio, nucleoli, and mild hyperchromasia. These changes are beyond those seen in reactive glandular epithelium; however, they fall short of the criteria for malignancy.
Changes suggestive of endocervical adenocarcinoma in situ include crowded hyperchromatic glandular cells in pseudostratified strips with occasional gland-like architecture or rosettes. Additionally, there can be peripheral feathering and prominent nucleoli. These features may be subtle, and the interpretation of adenocarcinoma in situ can be difficult. Challenging cases can be interpreted as atypical endocervical cells, favor neoplastic. Adenocarcinoma has more prominent malignant cytomorphologic features and commonly associated degenerated blood and necrosis. Adenocarcinoma can be endocervical, endometrial, or rarely metastatic in confirmatory tissue biopsy sections. Glandular and squamous abnormalities may be present in a single Pap test and each interpretation should be reported.
Publication of the 2019 ASCCP consensus guidelines in April 2020 introduced a change from test-result management to risk-based guidelines. The new guidelines change one management strategy for all with similar diagnoses and varied risk levels to patient management based on a combination of the patient’s level of risk, previous clinical history, and current screening test results. Risk levels from tables of risk variables from the 15-year Kaiser Permanente Northern California cervical cancer screening study were utilized for comparison to identify a clinical action threshold for patient management decisions (9, 35). Generally, patients at higher risk will undergo more frequent cervical carcinoma screening, followed by colposcopy and treatment as needed, while those at lower risk will have less frequent surveillance (35). Therefore, patients with similar Pap test results may be managed differently based on their risk for developing high-grade dysplasia.
In summary, the Pap test is a screening test for precancerous changes of the cervix. Screening intervals, management, and treatment are risk-based, taking into consideration the age of the patient, current cytology, pathology and HPV results, previous test results, age, and immune status.
A potential new approach in cervical cancer screening is the use of self-collected vaginal specimens for genotyping of HPV; however, self-sampling is not yet FDA-approved and is not currently the standard of care in the United States. Several studies have looked at the stability of these specimens, how these samples perform compared to clinician-collected samples, and advantages and potential concerns associated with this type of specimen.
Self-collected vaginal specimens for HPV genotyping are generally collected using a “dry” or lavage-based HPV self-sampling approach, most commonly using a brush/broom. A number of studies have evaluated recovery and stability of HPV DNA from exfoliated cervical cells attached to the hydrophobic material used for manufacture of the collecting brush/broom (36–39). In one study (39), HPV DNA stability was evaluated with exfoliated cells remaining on the brush/broom in a “dry” state with specimens stored at temperatures ranging from 4 to 30 °C for up to 32 weeks. At various time points, HPV genotyping was performed along with an assessment of the degree of DNA fragmentation in the combined extracted HPV and human genomic material. DNA fragmentation was modestly and progressively increased over time at all temperatures, however, HPV genotyping utilizing PCR demonstrated minimal increases in cycle threshold for oncogenic HPV genotypes.
The “gold standard” for evaluating the success of self-collected vaginal specimens for HPV screening is based on the correlation of HPV genotyping results obtained from self-collected specimens with those obtained from specimens collected by a trained clinician.
In the United States, utilization of self-collected vaginal specimens for HPV screening has thus far been relatively limited. Accordingly, the great majority of published studies using self-collected vaginal specimens have been conducted in foreign countries (40–45). Studies to examine the correlation between self-collected vaginal specimens and clinician-collected specimens for HPV screening were conducted in the Netherlands (16 410 total randomized patients) and in Mexico (25 061 total randomized patients), respectively.
In the Dutch study (40), 8212 participants were randomly allocated to the self-sampling group and 8198 to the clinician-based sampling group. 569 (7.4%) self-collected samples and 451 (7.2%) clinician-collected samples tested positive for HPV based on genotype analysis (relative risk 1.04 [95% CI, 0.92–1.17]). After a median follow-up duration for HPV-positive women of 20 months, the sensitivity and specificity of HPV testing did not differ between self-sampling and clinician-based sampling in terms of the detection of CIN 2 + or CIN 3 + lesions in the follow-up cytology testing. The authors concluded self-collected vaginal specimens for HPV genotyping could be used as a primary screening method in routine cervical cancer screening.
In the study from Mexico (41), 12 330 women were randomly assigned to the self-collected vaginal specimen arm and underwent HPV genotyping, with follow-up colposcopy on patients testing positive. An additional 12 731 patients were randomly assigned to undergo cervical cytology only. The goal was to determine whether self-collected vaginal specimen could identify patients with CIN 2 or worse as well as conventional cytology. HPV testing identified 117.4 women with CIN 2 or worse per 10 000 (95.2–139.5) compared with 34.4 women with CIN 2 or worse per 10 000 (23.4–45.3) identified by cytology. The relative sensitivity of self-collected vaginal specimens to identify CIN 2 or worse cervical cancer using HPV testing was 3.4 times greater (2.4-4.9) than cervical cytology alone. On the other hand, the positive predictive value of HPV testing for CIN 2 or worse was 12.2% (9.9-14.5) compared with 90.5% (61.7-100) for cytology alone. The authors concluded that despite the much lower positive predictive value for HPV testing of self-collected vaginal specimens compared with cytology, such testing might be preferred for detecting CIN 2 or worse in low-resource settings where restricted infrastructure reduces the effectiveness of cytology-based screening programs.
Additional, smaller-scale studies have largely supported the conclusions from these 2 pivotal studies (42, 43), and a detailed meta-analysis of self-collected vs clinician-collected samples was published for studies performed prior to 2014 (44). In addition, one study addressed self- vs clinician-collected specimens for HPV screening in post-menopausal women and demonstrated that, even in this population, there was no significant difference between the 2 sampling methods for extended HPV genotyping (P = 0.827) (45).
The benefits and potential drawbacks of self-collected vaginal specimens for HPV genotyping are summarized as follows:
Advantages (46–48)
Potential concerns (47)
The availability of screening, along with vaccination programs, has decreased the incidence and mortality rates of cervical cancer (49–51). Screening can detect precursors and early-stage disease of squamous cell carcinoma and adenocarcinoma. Treatment of precursors and early-stage disease can prevent the development of invasive cervical cancer and reduce cervical cancer mortality. The 3 available cervical screening strategies in the United States are (a) primary HPV screening, (b) co-testing with HPV testing and cervical cytology, and (c) cervical cytology alone.
Recommendations for screening aim to balance benefits of early detection of treatable lesions and reduction in incidence and mortality of cervical cancer with the potential risk of false positives, unnecessary procedures, and potential harms (e.g., patient discomfort, healthcare costs, and risks of treatment on future pregnancies). The most recent screening recommendations from the 2018 US Preventive Services Task Force (USPSTF) (52) and the 2020 American Cancer Society (ACS) (53) are detailed next and summarized in Table 2. The main differences between the 2 guidelines relate to age to initiate screening and the test used in individuals ages 21 to 29 years old.
The FDA approved the cobas® HPV test in March 2014 and the BD Onclarity™ HPV Assay in April 2018 for primary HPV testing for screening in individuals 25 years or older (54). Both of these tests are approved for partial HPV genotyping. It has been demonstrated that primary HPV screening is more effective than screening with cytology alone and performs similarly to and with lower costs than screening with co-testing (9, 55). The 2 FDA-approved tests for primary HPV screening are not available at all institutions. In many settings, co-testing will be ordered in lieu of primary testing until an FDA-approved primary test is available.
The USPSTF recommends that primary HPV testing not be used to screen individuals 21 to 29 years old as a stand-alone test. This is due to the high prevalence of HPV in those under the age of 30 (56, 57), although this may change as an increasing number of people are vaccinated. In one study, primary HPV screening starting at 25 years of age doubled the number of colposcopies but resulted in a 54% greater detection of CIN 3 + when compared to the same strategy starting at 30 years of age (58). However, despite the increased detection of CIN 3+, quick progression to cancer is uncommon, and so, on balance, cytology screening is felt to be adequate for detection of serious disease, while avoiding the potential for overevaluation associated with the highly sensitive HPV test in patients younger than 30 years old. Based on this data, the USPSTF recommends that primary HPV screening only be used for patients 30 years and older (52). An important difference in the ACS guideline is the recommendation for the use of primary HPV testing starting at 25 years old (53). Although based on the same data, the difference in interpretation reflects the balance of increased intervention (i.e., colposcopies) with increased number of precancerous lesions detected.
|
TABLE 2. Summary of screening recommendations |
||
|
US PREVENTIVE SERVICES TASK FORCE, 2018 |
AMERICAN CANCER SOCIETY, 2020
|
|
|
Age to start screening |
21 |
25 |
|
Age to end screening |
65 |
65 |
|
Screening test options and intervals |
Ages 21–65: Cytology alone every 3 years or Ages 21–29: Cytology alone every 3 years Ages 30–65: Cytology plus HPV testing every 5 years or Ages 21–29: Cytology alone every 3 years Ages 30–65: HPV testing alone every 5 years |
HPV testing alone every 5 years or Cytology plus HPV testing every 5 years or Cytology alone every 3 years |
|
Preferred strategies |
Cytology alone every 3 years and HPV testing alone every 5 years (equally preferred) among women ages 30–65 years. |
HPV testing alone every 5 years |
With regards to interval of screening, both organizations recommend screening with primary HPV not occur at intervals shorter than 3 years and not beyond 5 years among patients with negative screening results. An analysis by Ronco et al. concluded that a screening interval of at least 5 years for HPV screening is safer than cytology every 3 years (59).
In co-testing, cytology and HPV testing are collected and reported together. In addition to the two FDA-approved tests for primary HPV screening, the digene HC2 high-risk HPV DNA test, Cervista HPV HR assay, Cervista HPV 16/18 assay, Aptima HPV assay, and Aptima HPV 1618/45 assay are all approved by the FDA as of March 2019 for co-testing and are available at most institutions (54). As not all institutions currently have access to FDA-approved assays for primary HPV testing, providers may order co-testing when HPV-based testing is recommended. Depending on the clinical scenario, patient population, and shared-decision making with the patient, co-testing may be chosen by the provider if there is a concern for higher false-negative rates of cytology or HPV testing alone reported in the literature. As laboratories increase in capacity and access to FDA-approved primary HPV screening tests, either through new FDA-approvals or through switching to approved platforms, adoption of primary HPV testing may increase in alignment with the USPSTF and ACS preferred screening strategies.
The USPSTF recommends that co-testing be offered to patients 30 years and older with retesting in 5 years recommended after a negative screen (52). Similar to primary HPV testing, the ACS recommendation differs slightly in that cotesting is also acceptable among those older than 25 years old (53). The addition of HPV testing to cytology increases the detection of prevalent CIN 3 with a concomitant decrease in CIN 3 + or cancer detected in subsequent rounds of screening (60–62). The increase in diagnostic lead-time with co-testing translates into lower risk following a negative screen, which allows for an interval of 5 years between screens with incident cancer rates similar to or lower than screening with cytology alone at 3-year intervals (63, 64). The addition of HPV testing to cytology also enhances the identification of women with adenocarcinoma of the cervix and its precursors (64, 65). Compared to squamous cell cancers, cytology has been relatively ineffective in decreasing the incidence of invasive adenocarcinoma of the cervix (66).
When cervical cytology alone is used, the cervical sample is analyzed for cellular abnormalities. After cytology is performed, there is an option to perform reflex HPV testing when the cytology result returns positive for ASC-US. The USPSTF recommends screening for cervical cancer every 3 years with cervical cytology alone in women ages 21 to 29 years (52). The ASCCP recommends that, for patients ages < 25 years with ASC-US, reflex HPV testing be performed (7). Given the high prevalence of transient HPV infection among adolescents and young adults, initial screening at age 21 years should be with cytology alone. If cytology alone is used, the ACS recommends that the screening interval be every 3 years (53). Studies of screening intervals in women with a history of negative cytology results report an increased risk of cancer after 3 years even after controlling for prior number of negative cytology tests (67). Conversely, the incidence of high-grade cytology within 3 years of a normal cytology is low (10–66 per 10 000) (68) and modeling studies demonstrating that detection was similar with annual or triennial screening, but annual screening resulted in increased number of interventions (i.e., colposcopies) (69, 70).
There are no randomized trials comparing mortality rates among the various screening strategies. One modeling study found that HPV-based screening strategies (i.e., primary HPV testing or co-testing) were associated with fewer cervical cancer deaths (0.23–0.29 per 1000) compared with screening strategies that included cervical cytology (i.e., cytology alone or reflex HPV testing, 0.30–0.76 per 1000) (69).
With respect to detection, a systematic review found that primary HPV testing among individuals 25 to 65 years compared with cytology alone was associated with increased detection of CIN 3 + in the initial round of screening (relative risk range, 1.61 [95% CI, 1.09–2.37]) to 7.46 (95% CI, 1.02–54.66) (8). Colposcopy rates were higher for primary HPV testing than for cytology alone in one of 3 trials (NTCC Phase II) (62, 71) and similar in 2 trials [FINNISH (72) and HPV FOCAL (73)]. False-positive rates for CIN 2 + were higher for primary HPV testing alone than for cytology alone in one trial (NTCC Phase II) and similar in another trial (FINNISH).
In comparing detection of CIN 3 + using cotesting vs cytology alone, randomized control trials [NTCC Phase I (62, 71), SWEDESCREEN (60), POBASCAM (61), ARTISTIC (74)] have found that including HPV testing leads to earlier detection, but not reduced incidence, of high-grade cervical dysplasia and cancer. In all 4 trials, HPV testing in the first screening round detected cases of CIN 3 + that were missed by cytology, but there were fewer cases in the combined HPV testing plus cytology group at round 2, and over both screening rounds there were no significant differences. In contrast, the HPV FOCAL study found a lower incidence of CIN 3 + associated with initial HPV testing (incidence ratio 2.3 per 1000 [95% CI 1.5–3.5]) compared with initial Pap testing (incidence ratio 5.5 per 1000 [95% CI 4.2–7.2]); relative risk 0.42 [95% CI, 0.25–0.69]) (73). Colposcopy rates were higher for screening with co-testing than for cytology alone in 2 trials (ARTISTIC and NTCC Phase I) and not reported in the other 2 trials (SWEDESCREEN and POBASCAM). False-positive rates were higher for screening with co-testing in 3 of 4 trials (SWEDESCREEN did not report the false-positive rate for the intervention group).
A benefit of co-testing is that, among individuals with a negative co-test, the risk of developing CIN 3 + was less than 1% in the next 5 to 10 years (63, 64, 75–78). Meta-analysis indicated that, compared with cytology-based testing, screening with HPV testing (mainly with co-testing) was associated with a lower incident of cervical cancer at a median follow up of 6.5 years (rate ratio 0.60, 95%CI, 0.40– 0.89) (59). Consistent with the low risk associated with negative co-testing, modeling studies found that co-testing every 5 years was as effective as screening with cytology alone every 3 years (79) and was associated with decreased colposcopies compared with co-testing every 3 years, with only a minimal change in lifetime cancer risk (0.39% vs 0.61%) (80). Lastly, for centers with imaged LBC available, a recent study showed that more women subsequently diagnosed with cervical cancer within 1 year of co-testing were identified by the LBC results than by the HPV results (85.1%, 1015/1193 vs 77.5%, 925/1193), confirming the value of LBC element in co-testing (81).
Screening for cervical cancer in asymptomatic, immunocompetent patients, regardless of the age of sexual debut, should not be performed in individuals younger than 21 years old (53). Cervical cancer rates have been reported to be 0.15% in females 15 to 19 years old and 1.4% in women 20 to 24 years old (82). The prevalence of CIN 3 in women under 21 is estimated at 0.2% while the false-positive cytology rate is reported at 3.1%, emphasizing the potential harm of early screening (83, 84). This is because exposure of cervical cells to HPV during vaginal intercourse may lead to cervical precancers, but regression is common and is generally not a rapid process. Furthermore, screening initiation is not tied to sexual debut because, although the incidence of HPV infection is highest following the initiation of sexual intercourse, the infection usually clears spontaneously in 90% within 2 years (85). In counseling patients, it is important to emphasize the need for screening even after vaccination. This is because it is uncertain what level of vaccine uptake in the general population will achieve the level of individual protection and herd immunity that would warrant changes in screening protocols for all women or for those with documented vaccination history (53).
As noted, the USPSTF recommends screening at 21 years and older with cytology every 3 years (52) based on a meta-analysis of randomized trials and observational studies that demonstrated higher false-positive rates with HPV testing because of the higher rates of transient infection in this age group (8). Alternatively, the ACS recommends that screening begin at age 25 with primary HPV testing every 5 years (53). The higher age of screening initiation is based on the low incidence of cervical cancer (0.8%) due to high rates of spontaneous regression of HPV infection (53, 86–88). The ACS favors primary HPV testing based on randomized controlled trials showing higher sensitivity of HPV-based testing than cytology alone (59, 62), which is important in the context of increased vaccination rates (89). This will become increasingly relevant as a greater number of women are vaccinated prior to exposure to HPV.
The timing to discontinue screening depends on adequacy of screening, prior results, life expectancy, and patient preferences. Adequate screening is defined by (a) 2 consecutive negative HPV tests within the past 10 years (with the most recent within the previous 5 years), (b) 2 consecutive negative co-tests within the past 10 years (with the most recent within the previous 5 years), or 3 consecutive negative Pap tests within the past 10 years (with the most recent test within the previous 3 years) (53). If results for the past 10 years are unknown, screening would be considered inadequate. In addition to adequate screening, the patient should not have had CIN 2 or worse for the past 25 years.
The ACS and USPSTF both recommend that those over age 65 who have had regular screening in the past 10 years with normal results and no history of CIN2 + within the past 25 years discontinue screening (53). Those with a history of precancer or cancer should continue to have testing for at least 25 years after diagnosis even if the testing goes past age 65. The evidence for discontinuation of screening is based primarily on a single modeling study with a model of continued screening up to age 90 (69). A prolonged screening model only resulted in the reduction of 1.6 cancer cases and 0.5 cancer deaths per 1000 women compared to an additional 127 colposcopies per 1000 women. However, it is important to note that approximately 20% of cervical cancers occur in patients older than 65 years, and evidence indicates that screening in those 65 years and older is associated with a lower risk of subsequent development of cervical cancer (90, 91). In patients with inadequate prior screening or unknown screening history, the high incidence of mortality from cervical cancer and modeling studies suggest that screening older patients who have never been screened with cytology could reduce mortality by 74% (92–94). Based on this data, the USPSTF suggests, in those with inadequate or unknown prior screening, screening be continued until age 70 or 75 years old. Overall, data regarding the stopping age for screening are limited and should be based on an informed decision-making discussion with the patient.
Despite the somewhat nuanced differences between the ACS and USPSTF guidelines, there are 2 key concepts to the implementation of screening: (a) correctly identifying those who meet criteria for routine screening and (b) ensuring that patients who have abnormal Pap and/or HPV testing results are evaluated, usually by colposcopy with biopsy; undergo treatment if appropriate; and finally adhere to follow-up. Figure 1 includes these concepts and is adapted from the 2019 ASCCP guideline to demonstrate how a patient’s risk is evaluated, irrespective of which of the 3 screening strategies is used.
To determine if an individual meets criteria for routine screening, the following should be elicited from clinical history:
In summary, anyone with a history of immunosuppression, vulvar or vaginal dysplasia, hysterectomy with removal of cervix, clinical signs and symptoms, or prior abnormal results does not meet criteria for routine screening per the ACS or USPSTF guidelines. For those with abnormal prior results without recent testing, patients should be triaged based on the ASCCP guidelines described next and illustrated in Fig. 1.
The 2019 ASCCP risk-based management guidelines incorporate HPV testing and cytology results with prior test results to estimate an individual’s 5-year risk of CIN 3+ (9). The minimum amount of data required to generate a clinical action recommendation includes the patient’s age and current test results, recognizing that prior screening history might not be available. However, ideally, prior cytology, HPV and pathology data are entered into the risk calculator in order to create a personal risk score for the patient, which determines management. Data tables of risk estimates are to guide management clinical action thresholds under the principle of “equal management for equal risk” (9). The estimates are based on data from Kaiser Permanente Northern California (64), the BD Onclarity registrational trials (102, 103), the New Mexico HPV Pap Registry (104, 105), and the Centers for Disease Control and Prevention’s National Breast and Cervical Cancer Early Detection Program (106). Patients with an immediate risk of CIN 3 + that is less than 4% undergo surveillance, and based on their 5-year risk of CIN 3+, the interval may be 1, 3, or 5 years. Those with an immediate CIN 3 + risk of greater than or equal to 4% are referred to diagnostic evaluation, which may include colposcopic evaluation and/or excisional procedure.
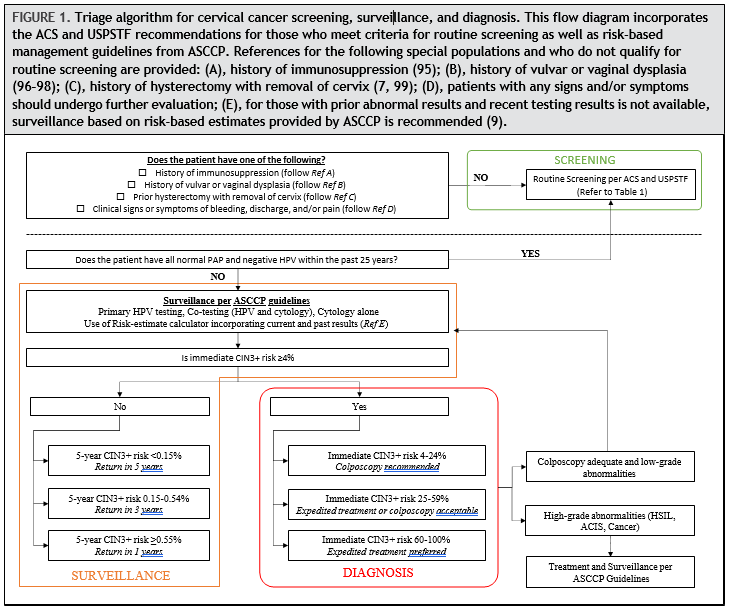
Surveillance is defined as follow-up testing at a shorter interval than that currently recommended for routine screening with either HPV primary testing or co-testing (i.e., sooner than 5 years). Surveillance is recommended for patients whose risk of CIN 3 + based on current test results and screening history is higher than the risk for the general screening population but lower than the risk at which colposcopy is recommended (7). For patients with an estimated 5-year CIN 3 + risk of less than 0.15%, return to routine screening at 5-year intervals using HPV-based testing is recommended. This is based on the estimated 5-year CIN 3 + risks after a negative HPV test (0.14%; 95% CI, 0.13%–0.15%) and co-test (0.12%; 95%CI, 0.12%–0.13%). Cytology alone is never recommended at 5-year intervals. For patients who have an estimated 5-year CIN 3 + risk of 0.15% or greater but less than 0.55%, repeat testing in 3 years with HPV-based testing is recommended. Finally, for those with an estimated risk of greater than 0.55% but less than 4% (threshold for immediate colposcopy), repeat testing in 1 year with HPV-based testing is recommended. For example, follow-up at 1 year is recommended after a screening test showing minimal abnormalities: HPV-positive/negative for intraepithelial lesion or malignancy or HPV-negative/LSIL with unknown previous screening history (immediate risks 2.1% and 1.1%, respectively) (9).
Surveillance also applies to patients who are referred for colposcopic evaluation and/or treatment and are found to have CIN 1 or normal results. The 5-year CIN 3 + risks for various clinical scenarios are available based on publicly available risk tables (https://CervixCa.nlm.nih.gov/ RiskTables). For individuals diagnosed with highgrade abnormalities and who are treated, more frequent surveillance with HPV-based testing at 6 months is preferred and, if positive, colposcopy with biopsies should be performed. Individuals treated for histologic HSIL with a subsequent abnormal screening test result have an elevated risk of cervical precancer warranting close follow-up (9, 107). HPV testing and co-testing are more sensitive than cytology alone in detecting CIN 2 + in both the post-colposcopy and post-treatment settings (108, 109).
The ASCCP guideline also addresses the issue of long-term follow-up surveillance after treatment for both high-grade and low-grade abnormalities (7). For those with a history of treated high-grade histology or cytology, after initial intensive surveillance period, the ASCCP recommends surveillance at 3-year intervals for at least 25 years, which may continue as long as the patient is in reasonably good health. This is based on data from long-term population studies that demonstrate a persistent 2-fold increase in cervical cancer risk after treatment of high-grade lesions (107). For those with history of low-grade cytology (HPV-positive negative for intraepithelial lesion or malignancy; ASC-US, or LSIL) or histologic LSIL abnormalities without evidence of histologic or cytologic highgrade, co-test in 1 year is advised, and, if results are all normal, they should be followed by continued surveillance at 3-year intervals.
Colposcopy standards have been outlined by the ASCCP (110, 111). It is recommended that practitioners follow the standardized terminology, which captures 6 major areas: (a) general assessment, (b) evaluation for presence of any acetowhite lesions, (c) description of normal colposcopic findings, (d ) description of abnormal colposcopic findings, (e) description of other/miscellaneous findings, and (f ) reporting of the colposcopic impression, defined as the highest grade impression of any visible lesion on the cervix. A comprehensive colposcopic examination should include description of the cervix visibility, squamocolumnar junction visibility, presence of acetowhitening, presence and visualization of a lesion, color/contours/borders/vascular changes of lesions, the location and size(s) of lesion(s), other features, and the colposcopic impression. A diagram or marked image annotating the findings should also be included. Minimum criteria for reporting findings at colposcopic examination should include the following: squamocolumnar junction visibility (fully/not fully), acetowhitening (yes/no), lesion(s) present (acetowhite or other) (yes/no), and colposcopic impression (normal/benign, low grade, high grade, cancer). Colposcopy training is currently not regulated in United States, and there is no certification (112). Standards in many other countries do include training and generally stipulate that all clinicians who perform colposcopic examinations should have completed a formal colposcopic training program conducted by expert trained personnel whose clinical competence and teaching abilities are well documented (113).
For those at lowest risk (i.e., less than HSIL cytology, no evidence of HPV 16/18 infection) with a completely normal colposcopic impression, random biopsies are not recommended. This is based on Kaiser Permanente Northern California data that demonstrated that the risk of occult CIN 2 + was 1% to 7% and CIN 3 + was less than 1% in the afore-described low-risk group, which underwent 4-quadrant biopsies and endocervical curettage in that cohort. If these criteria are not met, multiple targeted biopsies (at least 2 and up to 4) are recommended, targeting all acetowhite areas to improve detection of precancers. Moreover, biopsies are needed for any degree of acetowhitening, metaplasia, or abnormalities (111).
In nonpregnant women 25 years and older with a very high risk of precancer, either immediate excisional treatment without biopsy confirmation or colposcopy with multiple targeted biopsies is acceptable (111). High risk in this context is defined as at least 2 of the following: HSIL cytology, HPV16 and/or HPV 18 positive, high-grade colposcopy impression. This is based on systematic review of see-and-treat management strategies for patients meeting the high-risk criteria, which found that 89% of all women with HSIL had CIN 2+, whereas other studies have shown somewhat lower risk from 73% to 86% (111, 114, 115). Endocervical curettage is preferred for nonpregnant patients when colposcopy is inadequate and in those not at lowest risk and no lesion is identified. It can also be considered when a lesion is seen (116).
HPV induces histologic changes in the squamous epithelium of the uterine cervix, particularly at the transformation zone. These changes comprise a diverse spectrum of alterations (Fig. 2). On one end of the spectrum are mild koilocytic changes, which have a degree of overlap with reactive atypia. On the other end of the spectrum are atypical basaloid epithelial cells involving the full thickness of a markedly thickened squamous epithelium. Lesions along this spectrum must be classified into discrete categories to guide clinical management. Two schemata are current recognized to do this: the 3-tier CIN system and the 2-tier SIL (1). The CIN system classifies lesions as CIN 1, 2, or 3, ranging lowest to highest grade. CIN 1 includes lesions with koilocytic changes and basal atypia confined to the lower one-third of the epithelial thickness. CIN 2 includes lesions with basal atypia involving the lower and middle thirds of the epithelial thickness. CIN 3 includes those with full-thickness basal atypia. The SIL system classifies lesions as either high grade (HSIL) or low grade (LSIL). LSIL includes CIN 1. HSIL includes CIN 2 and CIN 3. While the latter category has the benefit of simplicity, it loses the informative distinction between CIN 2 and CIN 3.
There is only moderate reproducibility among pathologist in classifying HPV-induced squamous lesions, using both the CIN and SIL systems. This is largely a consequence of the great diversity in the histomorphology of these lesions and the substantial fraction of cases with features that are not clearly high or low grade. For example, while reproducibility is good for the distinction between CIN 1 and CIN 3 (117, 118), reproducibility is poor for the diagnosis of CIN 2 (117, 119), which is often difficult to distinguish from CIN 1 and CIN 3. Consistency in diagnosis has been aided by the addition of immunohistochemistry for p16, a protein product of the cell cycle gene CDKN2A. This marker is sensitive for high-grade lesions but is also expressed in a substantial subset of lowgrade lesions. Expression of p16 is particularly high in low-grade lesions driven by high-risk HPV types, with diffuse expression of p16 seen in nearly 90% of hrHPV-positive LSIL in one study (120). CIN 1 lesions that are p16-positive progress to CIN 2 or higher in 10% to 35% of cases, while those that are p16-negative progress in <5% of cases (118). The negative predictive value of p16 is thus high for predicting progression to a highgrade squamous lesion.
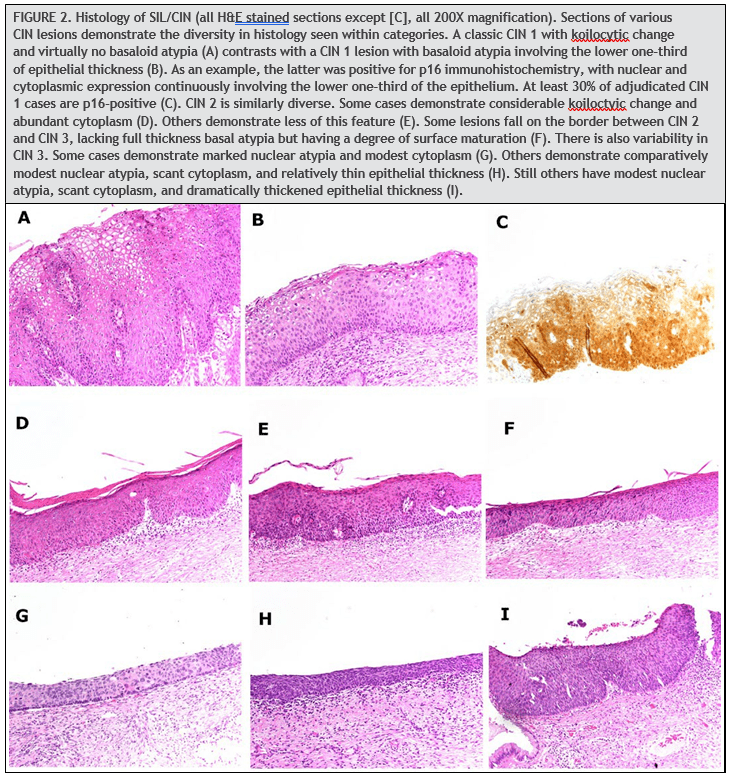
The Lower Anogenital Squamous Terminology guidelines by the College of American Pathologists and the ASCCP recommend using p16 immunohistochemistry when the differential is between precancer (CIN2/3) and a mimic of precancer (121). In addition, if the pathologist is entertaining an H&E morphologic interpretation of CIN 2, p16 immuno-histochemistry is recommended to help clarify the situation. Strong and diffuse block-positive p16 results support a categorization of precancer. Negative or non-block-positive staining strongly favors an interpretation of low-grade disease or a non–HPV-associated pathology (121). The Lower Anogenital Squamous Terminology guidelines recommend against the use of p16 as a routine adjunct to histologic assessment of biopsy specimens with morphologic interpretations of negative, CIN 1, and CIN 3. However, in special circumstances, p16 may be used as an adjunct to morphologic assessment for biopsy specimens interpreted as CIN 1 that are at high risk for missed high-grade disease, which is defined as a prior cytologic interpretation of HSIL, ASC-H, ASC-US/HPV-16þ, or AGC (not otherwise specified) (121). Positivity for p16 is defined specifically as continuous strong nuclear or nuclear plus cytoplasmic staining of the basal cell layer with extension upward involving at least one-third of the epithelial thickness (Fig. 2C).
Based on the previous discussion of the importance of specifying the indication for testing (i.e., screening, surveillance, or diagnosis) and the test used, we propose the following report template (Table 3) to facilitate results interpretation and clinical decision-making. While this template can be modified for local needs, we believe it incorporates the most important components. It is important to allow for all available (or most recent) prior results to be summarized in the current report to facilitate risk-based decision-making. Furthermore, the specific HPV test used by the laboratory should be specified. Note the p16/Ki67 dual-stain may be performed in cases where cytology results are abnormal (LSIL or ASCUS) and/ or hrHPV-positive, but it has not been included in the current guidelines and is optional (122).
The goal of a screening protocol is to optimize the detection of precancerous lesions at a time when they are treatable while limiting the harm of overtreating benign disease. This begins with correctly identifying those patients suitable for routine screening vs those who require surveillance and/or diagnosis. The introduction of risk-based management considers factors that influence clinical action thresholds allowing for greater tailoring of screening strategy for patients. The most recent ASCCP guideline highlights that prior history profoundly influences risk estimates, specifically current HPV and cytology test results, previous HPV test results, and history of histologic HSIL (9). The estimated risk guides decisions regarding surveillance interval, colposcopic referral, and treatment. In all 3 recommendations, the concepts of screening, surveillance, and diagnosis are important in framing the clinical situation at hand and the appropriate use and interpretation of tests. For example, the intervals of 1-, 3-, and 5-year discussed within the ASCCP guidelines are surveillance intervals whereas the 3- and 5- year intervals discussed in the ACS and USPSTF guidelines refer to screening intervals. Furthermore, the ACS and USPSTF guidelines were developed prior to the ASCCP guidelines, and nuanced differences may be noted, specifically with updates to the use of primary HPV testing. For example, the ASCCP guidelines recommend that when primary HPV screening is used as the initial test alone, additional reflex triage test (e.g., reflex cytology) for all positive HPV tests be performed regardless of genotype (7); this is a change from the 2015 interim guidelines (58). However, if primary HPV screening test genotyping results are HPV 16- or HPV 18-positive and reflex triage testing from the same laboratory specimen is not feasible, patients should proceed directly to colposcopy (58). The perspective of the ASCCP guidelines is to use surveillance to address potential clinical situations involving abnormal results (e.g., HPV-positive) whereas the ACS and USPSTF guidelines target routine screening in lowrisk patients. Lastly, once an individual has an abnormal test result, depending on subsequent findings and estimated risk, the majority will remain in surveillance with a small subset who would qualify to return to routine screening.
|
TABLE 3. HPV and cervical cancer testing report template. |
|||
|
Patient identification |
Name |
||
|
Date of birth |
|||
|
Medical record number |
|||
|
Date of collection |
|||
|
Accession number |
|||
|
Name of submitting physician and/or clinic |
|||
|
Indication |
|
||
|
|||
|
|||
|
Clinical history |
Provider description |
||
|
Pregnant? â– Yes â– No |
|||
|
Immunosuppressed? â– Yes â– No |
|||
|
Prior results |
Date |
Result |
|
|
Cytology |
|||
|
HPV |
|||
|
Histopathology |
|||
|
Current testing |
|
||
|
|||
|
|||
|
Current results |
HPV |
|
|
|
|||
|
HPV test used |
|||
|
HPV genotype (if positive) |
|
||
|
|||
|
|||
|
|||
|
Cytology |
Per Bethesda terminology |
||
|
p16/Ki67 dual-stain |
|
||
|
|||
|
|||
|
Other adjunctive tests (please specify) |
|||
|
Name of reviewing pathologist |
|||
|
Date of report |
|||
|
Name and address of the laboratory |
|||
Moving forward, several future directions in research and implementation have the potential to improve access and implementation of these guidelines. Given that the risk estimates are based on both current and prior testing results, automated extraction from medical records and laboratory reports would simplify risk-estimate calculation. Ideally, standardized reports would include HPV test used, genotype information, cytology, and histology using common terminology (e.g., Lower Anogenital Squamous Terminology) integrated with other clinical information from a patient’s electronic health record. This would not only allow for accurate risk estimates but also establish a reliable tracking and reminder system to facilitate communication, improve patient safety and quality of care, and minimize missed or delayed diagnoses. Second, as additional HPV tests and data from studies become available, the FDA assessment of HPV assays may potentially increase the number of tests approved for primary HPV testing. Primary HPV testing is attractive as it has been demonstrated to be more effective than screening with cytology alone and performs similarly to and with lower costs than screening with co-testing. In addition, HPV testing is also more amenable to self-collection, which opens new opportunities to screen difficult to reach and underscreened populations at high risk of cervical cancer (123–125). Ultimately, the key message to patients, and providers alike, is stated by the ACS: “The most important thing to remember is to get screened regularly, no matter which test you get.”
Nonstandard Abbreviations: ACS, American Cancer Society; ASCCP, American Society for Colposcopy and Cervical Pathology; AGC, atypical glandular cells; ASC-H, atypical squamous cells—cannot exclude high-grade squamous intraepithelial lesion; ASC-US, atypical squamous cells of undetermined significance; CIN, cervical intraepithelial neoplasia; FDA, Food and Drug Administration; HSIL, high-grade squamous intraepithelial lesion; hrHPV, high risk human papilloma virus; HPV, human papilloma virus; LBC, liquid-based cytology; LSIL, low-grade squamous intraepithelial lesion; USPSTF, US Preventative Services Task Force.
Author Contributions: The corresponding author takes full responsibility that all authors on this publication have met the following required criteria of eligibility for authorship: (a) significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data; (b) drafting or revising the article for intellectual content; (c) final approval of the published article; and (d) agreement to be accountable for all aspects of the article thus ensuring that questions related to the accuracy or integrity of any part of the article are appropriately investigated and resolved. Nobody who qualifies for authorship has been omitted from the list.
Authors’ Disclosures or Potential Conflicts of Interest: Upon manuscript submission, all authors completed the author disclosure form. Disclosures and/or potential conflicts of interest: Employment or Leadership: S.O.A. Leung, American College of Obstetricians and Gynecologists, District I Quebec Section Fellow Vice Chair, International Papillomavirus Policy Committee Member, American Cancer Society Cervical Cancer Screening Initiative, Provider Needs Workgroup Member; S. Feldman, Board of IPVS. Consultant or Advisory Role: None declared. Stock Ownership: None declared. Honoraria: S. Feldman, Indian Health Service, post-graduate courses at Harvard Medical School. Research Funding: Y. Zhu, funding from Abbott Laboratories to institution; M.H. Creer, Abbott Clinical Diagnostics, Siemens Clinical Diagnostics, Fujirebio; S. Feldman, funding from National Cancer Institute/National Institutes of Health and Society to Improve Diagnosis in Medicine to institution, gift to institution from Glyciome. Expert Testimony: None declared. Patents: None declared. Other Remuneration: S. Feldman, royalties from Uptodate, payment for travel from American Cancer Society and ASCCP.
Role of Sponsor: The funding organizations played no role in the design of study, choice of enrolled patients, review and interpretation of data, preparation of manuscript, or final approval of manuscript.
Guidance Documents are brought to you by the