
Advocacy - Comment Letter
Dear Chair Sanders, Ranking Member Cassidy, Chair McMorris Rodgers, and Ranking Member Pallone:
The Association for Diagnostics and Laboratory Medicine (ADLM) is concerned about the impact of the Food and Drug Administration’s (FDA’s) May 6, 2024 final rule regulating laboratory developed tests (LDTs) on the ability of clinical laboratories to provide timely, quality patient care. Over the past few months, the association has conducted several surveys to get input from these front-line laboratories regarding how FDA oversight would affect their ability to serve their patients.
Our first survey, which was conducted in April 2024, prior to the release of the final rule, indicated that:
- Hospitals were worried that duplicative FDA oversight would limit patient access to testing services and delay the delivery of care; and
- Hospitals did not believe that FDA oversight would improve the quality of the testing they provided, but instead would force them to reduce the services they offer due to financial strain.
Our second survey, which was conducted in June 2024, after the publication of the FDA final rule, reported that:
- Laboratories were concerned they did not have the staff or resources to meet the requirements set forth for previously marketed (i.e., grandfathered) LDTs under the final rule, with more than one-half of the testing facilities stating they would discontinue some LDTs.
- Similarly, most laboratories responded they would be unable to comply with the FDA’s requirements associated with the unmet need exceptions with 60% of the hospitals stating they would be unable to comply with the requirements associated with the exception and nearly 70% of those labs stating they would discontinue LDTs.
Our third survey, which was conducted in July and early August 2024, continued to explore the impact of the LDT final rule on clinical laboratories and their ability to provide testing services. Laboratories were specifically asked about the premarket review provisions, potential delays in patient care, and which agency should regulate LDTs.
Pre-Market Review
The survey asked participants (121 respondents) about the FDA provision that allows laboratories to forego agency review if their LDTs are approved by the New York’s Department of Health’s Clinical Laboratory Evaluation Program. While the rule exempts labs from FDA pre-market review, they remain subject to FDA labeling, registration, medical device reporting, and inspection requirements, in addition to other FDA provisions and the existing CLIA standards.
Laboratories were asked a series of questions:
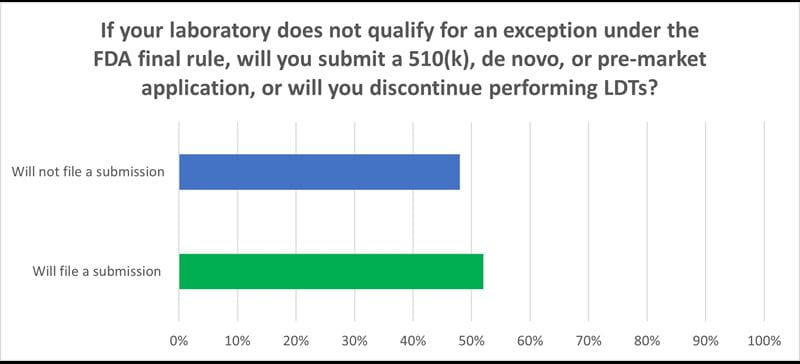
- If you do not qualify for an exception under the FDA rule will you seek approval for your LDTs or discontinue them.
- Forty eight percent of the laboratories responded they would discontinue their LDTs. This is a significant finding. If, according to the FDA, there are approximately 12,000 laboratories qualified to do LDTs in the United States (out of more than 300,000), nearly 6,000 will stop offering these critical tests.
- For those who stated they would seek approval for their tests, a slight majority chose NYS. However, more than 80 percent of the laboratories stated they have never filed a submission, which means they are likely to need significant help gathering and submitting the data and incur sizeable costs.
- Nearly 70 percent of the laboratories are concerned that NYS will not accept applications from those laboratories not performing testing on in-state residents.
Delay Assess to Care
One of the concerns within the healthcare and laboratory communities is that the FDA rule may result in many laboratories discontinuing their LDTs resulting in a delay of testing services. Laboratories were asked (159 respondents) if they stopped performing their LDTs, was there another laboratory within their state to offer the test(s).
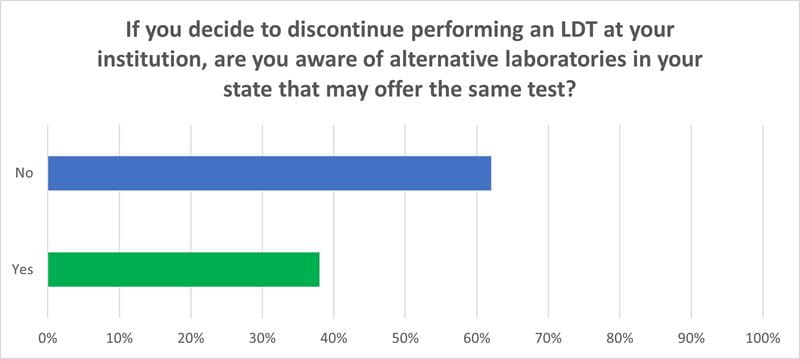
- More than 60 percent of the laboratories responded that there is no alternative laboratory within their state to offer the LDTs they perform. If this occurs, it will create a hardship for many individuals, particularly among marginalized communities, and those living in rural areas, who do not have the means to travel to another region of the country to obtain the test needed to diagnose and treat their condition.
Agency Oversight
A final wrap-up question asked participants (170 responses) if additional LDT oversight is necessary, which government agency should provide it – the FDA or CMS/CLIA.
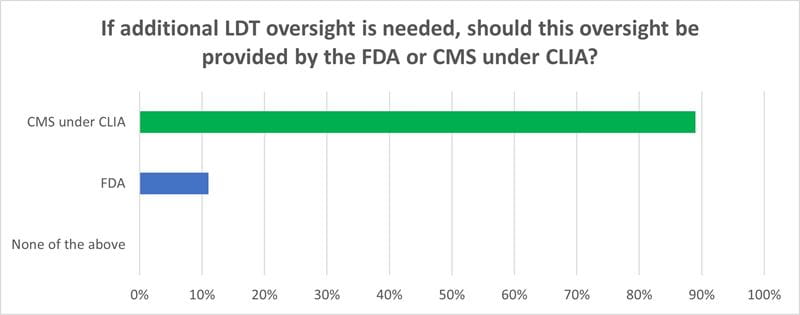
An overwhelming majority, 89 percent, chose CMS and CLIA. This response likely reflects the long-time relationship between CMS and the laboratory community, which dates back to 1965. Thus, while clinical laboratories are familiar with CMS operations and terminology, they are not familiar with the FDA as the agency regulates medical device manufacturers and test kits, not testing facilities.
ADLM urges lawmakers to rescind the FDA rule, while courts determine whether the agency has the legislative authority to regulate these tests. Concurrently, we encourage Congress to discuss separate remedies to streamline the FDA review process and review and update CMS standards pertaining to LDTs. ADLM looks forward to working with you on these most important issues. If you have any questions, please email Vince Stine, PhD, ADLM’s Senior Director of Government and Global Affairs, at [email protected].
Sincerely,
Anthony A. Killeen, MD, MSc, PhD
President, ADLM